Structural Biology, Molecular Interaction Modeling, Crystal Engineering
The BioMIMIC team is a multidisciplinary group of experts with backgrounds in both physics and biology. By combining their knowledge and working together, they aim to achieve a thorough understanding of molecular systems at the atomic scale. This diversity and collaboration enable the team to carry out innovative research in Structural Biology, Molecular Interaction Modelling, and Crystal Engineering.
To achieve this research, the team employs a combination of internationally recognized innovative methods, such as the use of charge density analysis derived from quantum chemical calculations or obtained from diffraction experiments, i.e. ultra-high resolution crystallography. The accurate determination of these properties is crucial, as electrostatic forces play a key role in molecular recognition and assembly, and biochemical reactions. The team also uses a combination of the topological analyses of the electron density, the Laplacian of the electron density and the electrostatic potential, as well as the topography of the electric field lines, to characterize intermolecular interactions in terms of both orientation and strength and to describe the influence zones of molecular electrophilic and nucleophilic sites in the space. These developments provide valuable tools for analyzing interactions, such as the sigma-hole interaction, which is directly linked to molecular reactivity. The team also utilizes established techniques, such as NMR, X-ray diffraction, and in silico modelling, to study the structure-function relationships of enzymes and structural proteins.
Head of
- DIDIERJEAN Claude, University professor
Members
- AUBERT Emmanuel, Associate professor
- BEGUE Dunkan, PhD Student
- ESPINOSA Enrique, Laboratory Director and University professor
- FAVIER Frédérique, Associate professor
- GOMES Antoniel AS, Postdoc
- GUILLOT Benoît, University professor
- JELSCH Christian, CNRS research director
- MAMBATTA Haritha, Postdoc
- MARQUES LEITE Miguel, PhD Student
- MATHIOT Sandrine, Technician
- PONSARD Bastien, Fixed-term contract engineer
- TSAN Pascale, Associate professor
Structure-function relationships
We are recognized for our ability to experimentally obtain three-dimensional structures of biological macromolecules and to analyze their structure function relationships. A first research topic on which we have more than 10 years of expertise concerns glutathione transferases and their complexes with small ligands.
A second one is focused on the characterization, at the molecular level and from a fundamental point of view, of the bacterial conjugation allowing these microorganisms to evolve rapidly, for example in order to resist an antibiotic.
We also have an expertise in the analysis of the conformations of peptidomimetics in the solid state.
We determine the structures by X-ray crystallography or by NMR. Our crystallogenesis platform includes two pipetting robots and a semi-automatic binocular. It allows us to perform many tests and increases the chances of obtaining crystals. Thanks to the X-ray diffraction and scattering platform hosted by the laboratory, we can perform preliminary tests of the crystals at home, and even collect experimental data necessary to establish a preliminary structure. Our access to the SOLEIL and ESRF synchrotrons allows us to improve them at maximum resolution.
Molecular modelling at ultra high resolution
An extensive survey of Cambridge Structural Database is carried out to study directionality and stereochemistry of hydrogen bonds with an oxygen acceptor including carbonyl, alcohols, phenols, ethers and esters groups. The results obtained through this survey are correlated with the charge density of these different chemical groups. The electron density of these different oxygen atoms types show striking dissimilarities in the electron lone pairs configuration.
Crystal Enginering
The Crystal Engineering group focuses on the control of both molecular organization in space and modulation of the intensity of the intermolecular interactions involved in this organization. The study of directional intermolecular interactions is at the center of our approach. It relies on the use of the influence of intra- and intermolecular environment in order to drive the crystalline molecular arrangement, and thus to modify the physicochemical properties of the crystalline solids. The study of the electron distribution and its derived properties from experimental (XRD) and theoretical (quantum calculations) methods allows us to characterize the interatomic and intermolecular interactions involved on.
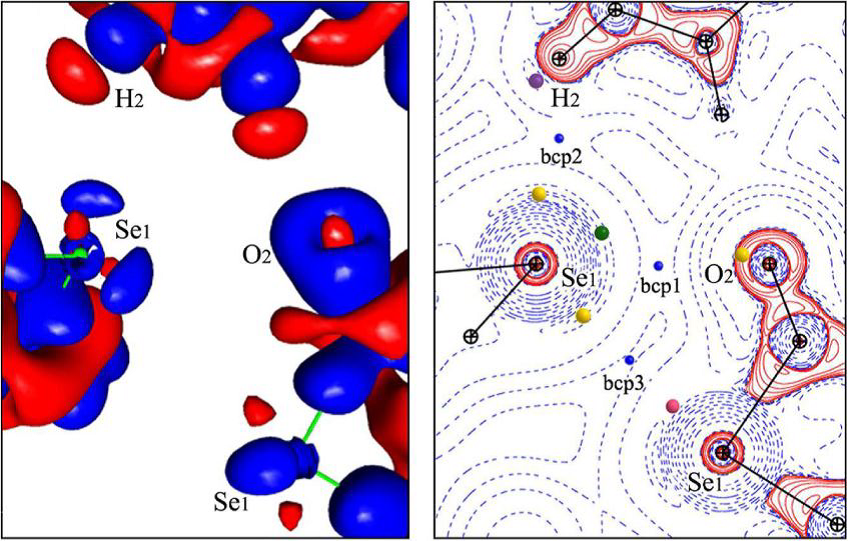
Software MoProSuite
The refinement program MoPro (Molecular Properties) is dedicated to the refinement at (sub)atomic resolution of structures ranging from small molecules to biological macromolecules. MoPro uses the multipolar pseudo-atom model for the electron-density refinement. Alternative methods are also proposed, such as modelling bonding and lone-pair electron density by virtual spherical atoms.