
Vuković, V., Leduc, T., Jelić-Matošević, Z., Didierjean, C., Favier, F., Guillot, B., & Jelsch, C. (2021). A rush to explore protein–ligand electrostatic interaction energy with Charger. Acta Crystallogr. D77(10).
The mutual penetration of electron densities between two interacting molecules complicates the computation of an accurate electrostatic interaction energy based on a pseudo-atom representation of electron densities. The numerical exact potential and multipole moment (nEP/MM) method is time-consuming since it performs a 3D integration to obtain the electrostatic energy at short interaction distances. Nguyen et al. [(2018), Acta Cryst. A74, 524–536] recently reported a fully analytical computation of the electrostatic interaction energy (aEP/MM). This method performs much faster than nEP/MM (up to two orders of magnitude) and remains highly accurate. A new program library, Charger, contains an implementation of the aEP/MM method. Charger has been incorporated into the MoProViewer software. Benchmark tests on a series of small molecules containing only C, H, N and O atoms show the efficiency of Charger in terms of execution time and accuracy. Charger is also powerful in a study of electrostatic symbiosis between a protein and a ligand. It determines reliable protein–ligand interaction energies even when both contain S atoms. It easily estimates the individual contribution of every residue to the total protein–ligand electrostatic binding energy (Figure 1). Glutathione transferase (GST) in complex with a benzophenone ligand was studied due to the availability of both structural and thermodynamic data. The resulting analysis highlights not only the residues that stabilize the ligand but also those that hinder ligand binding from an electrostatic point of view. This offers new perspectives in the search for mutations to improve the interaction between the two partners. A proposed mutation would improve ligand binding to GST by removing an electrostatic obstacle, rather than by the traditional increase in the number of favourable contacts.
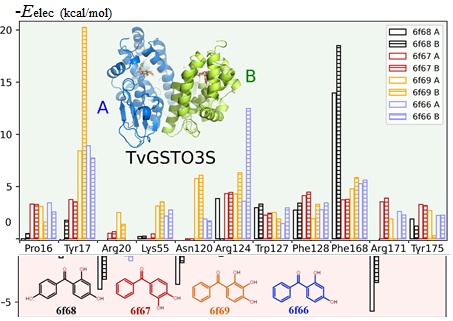
Figure 1.
Negative electrostatic (without polarization) interaction energy contributions of individual residues to GST-ligand binding for the two GST monomers (A and B). Positive numbers indicate favourable binding contribution (green background), and negative numbers indicate unfavourable binding (red background).
Leduc, T., Aubert, E., Espinosa, E., Jelsch, C., Iordache, C., & Guillot, B. (2019). Polarization of Electron Density Databases of Transferable Multipolar Atoms. The Journal of Physical Chemistry A, 123(32), 7156-7170.
Polarizability is a key molecular property involved in either macroscopic (i.e., dielectric constant) and microscopic properties (i.e., interaction energies). In rigid molecules, this property only depends on the ability of the electron density (ED) to acquire electrostatic moments in response to applied electric fields. Databases of transferable electron density fragments are a cheap and efficient way to access molecular EDs. This approach is rooted in the relative conservation of the atomic ED between different molecules, termed transferability principle. The present work discusses the application of this transferability principle to the polarizability, an electron density-derived property, partitioned in atomic contributions using the Quantum Theory of Atoms In Molecules topology. The energetic consequences of accounting for in situ deformation (polarization) of database multipolar atoms are investigated in detail by using a high-quality quantum chemical benchmark.
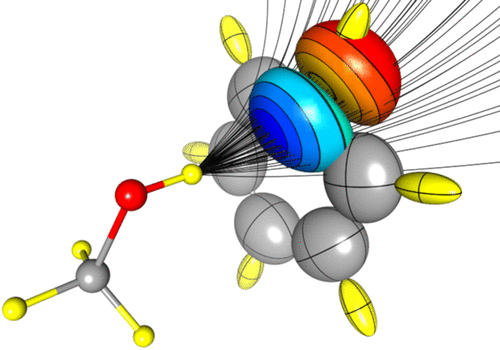
Figure 2.
Atomic polarizabilities tensors represented as ellipsoids.
Fournier B., Guillot B., Lecompte C., Escudero-Adan E. C. & Jelsch C. (2017). Advanced tools for charge density refinement and estimation of errors. Acta Crystallogr. A73, C569-C569.
Estimating uncertainties of property values derived from a charge-density model is not straightforward. A methodology, based on calculation of sample standard deviations (SSD) of properties using randomly deviating charge-density models, is proposed with the MoPro software. The parameter shifts applied in the deviating models are generated in order to respect the variance–covariance matrix issued from the least-squares refinement. This `SSD methodology’ procedure can be applied to estimate uncertainties of any property related to a charge-density model obtained by least-squares fitting. This includes topological properties such as critical point coordinates, electron density, Laplacian and ellipticity at critical points and charges integrated over atomic basins. Errors on electrostatic potentials and interaction energies are also available now through this procedure (Figure 3). The method is exemplified with the charge density of compound (E)-5-phenylpent-1-enylboronic acid, refined at 0.45 Å resolution. The procedure is implemented in the freely available MoPro program dedicated to charge-density refinement and modelling.
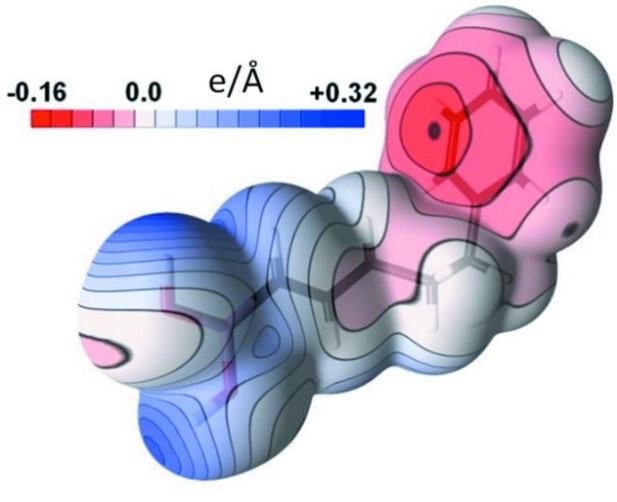
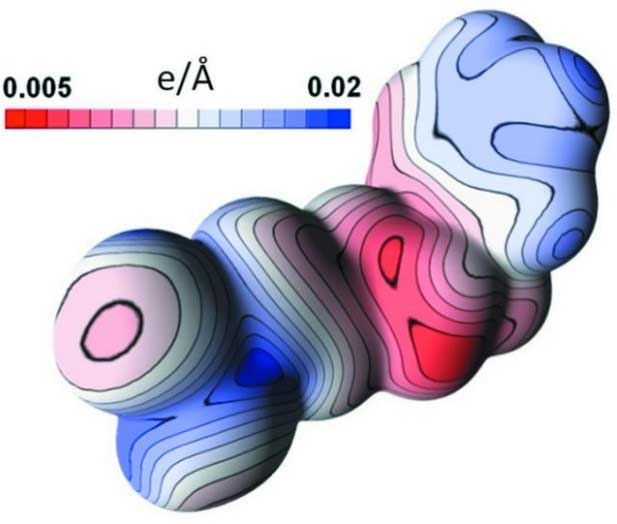
Figure 3.
Electrostatic potential on the molecule surface (left) and its standard deviation (right)
Jelsch, C., & Bibila Mayaya Bisseyou, Y. (2017). Atom interaction propensities of oxygenated chemical functions in crystal packings. IUCrJ, 4(2), 158-174.
The crystal contacts of several families of hydrocarbon compounds substituted with one or several types of oxygenated chemical groups were analyzed statistically using the Hirshfeld surface methodology. The propensity of contacts to occur between two chemical types is described with the contact enrichment descriptor. The systematic large enrichment ratios of some interactions like the O—H⋯O hydrogen bonds suggests that these contacts are a driving force in the crystal packing formation (Figure 4). The same statement holds for the weaker C—H⋯O hydrogen bonds in ethers, esters and ketones, in the absence of polar H atoms. The over-represented contacts in crystals of oxygenated hydrocarbons are generally of two types: electrostatic attractions (hydrogen bonds) and hydrophobic interactions. While Cl⋯O interactions are generally avoided, in a minority of chloro-oxygenated hydrocarbons, significant halogen bonding does occur. General tendencies can often be derived for many contact types, but outlier compounds are instructive as they display peculiar or rare features. The methodology also allows the detection of outliers which can be structures with errors. For instance, a significant number of hydroxylated molecules displaying over-represented non-favorable oxygen–oxygen contacts turned out to have wrongly oriented hydroxyl groups. Beyond crystal packings with a single molecule in the asymmetric unit, the behavior of water in monohydrate compounds and of crystals with Z′ = 2 (dimers) are also investigated. It was found in several cases that, in the presence of several oxygenated chemical groups, cross-interactions between different chemical groups (e.g. water/alcohols; alcohols/phenols) are often favored in the crystal packings. While some trends in accordance with common chemical principles are retrieved, some unexpected results can however appear. For example, in crystals of alcohol–phenol compounds, the strong O—H⋯O hydrogen bonds between two phenol groups turn out to be extremely rare, while cross contacts between phenols and alcohols have enriched occurrences.
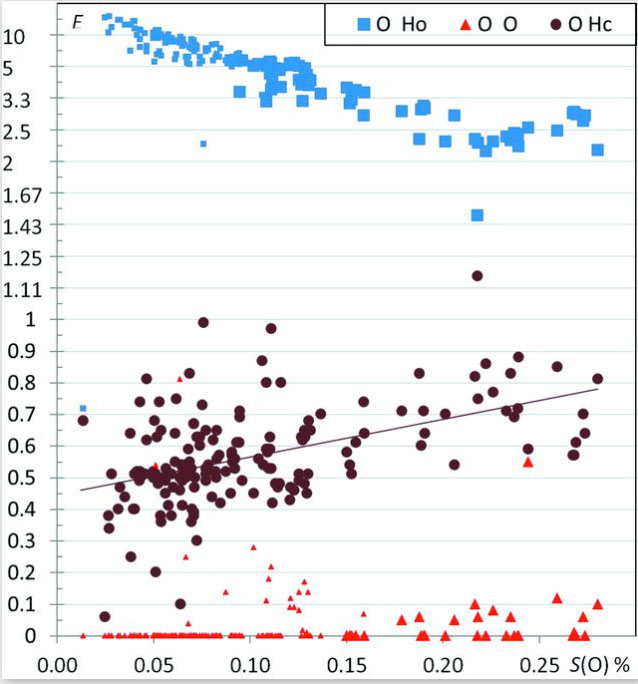
Figure 4.
Contact enrichment ratios in crystals of alcohols as a function of SO, the percentage of oxygen on the Hirshfeld surface. Ho and Hc refer to the hydrogen bound to oxygen and carbon atoms, respectively. Small symbols correspond to cases where the equiprobable contacts Rxy are smaller than 2%.
Selection of references: (see open access versions on https://hal.archives-ouvertes.fr/)
Vuković, V., Leduc, T., Jelić-Matošević, Z., Didierjean, C., Favier, F., Guillot, B., & Jelsch, C. (2021). A rush to explore protein–ligand electrostatic interaction energy with Charger. Acta Crystallographica Section D: Structural Biology, 77(10).
Guillot, B., Jelsch, C., & Macchi, P. (2021). Multipole modeling with MoPro and XD. Book chapter #10. In Complementary Bonding Analysis (pp. 235-268). De Gruyter.
Jha, K. K., Gruza, B., Chodkiewicz, M. L., Jelsch, C., & Dominiak, P. M. (2021). Refinements on electron diffraction data of β-glycine in MoPro: a quest for an improved structure model. Journal of Applied Crystallography, 54(4).
Leduc, T., Aubert, E., Espinosa, E., Jelsch, C., Iordache, C., & Guillot, B. (2019). Polarization of Electron Density Databases of Transferable Multipolar Atoms. The Journal of Physical Chemistry A, 123(32), 7156-7170.
Gallagher, J. F., Farrell, M., Hehir, N., Mocilac, P., Aubert, E., Espinosa, E., … & Jelsch, C. (2019). At the Interface of Isomorphous Behavior in a 3× 3 Isomer Grid of Monochlorobenzamides: Analyses of the Interaction Landscapes via Contact Enrichment Studies. Crystal Growth & Design, 19(11), 6141-6158.
Jelsch, C., Guillot, B., Leduc, T., & Fournier, B. (2017). Advanced tools for charge density refinement and estimation of errors. In Acta Crystallographica A. 73, C569-C569.
Jelsch, C., & Bibila Mayaya Bisseyou, Y. (2017). Atom interaction propensities of oxygenated chemical functions in crystal packings. IUCrJ, 4(2), 158-174.
Nassour, A., Domagala, S., Guillot, B., Leduc, T., Lecomte, C., & Jelsch, C. (2017). A theoretical-electron-density databank using a model of real and virtual spherical atoms. Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials, 73(4), 610-625.
Devi, R. N., Jelsch, C., Israel, S., Aubert, E., Anzline, C., & Hosamani, A. A. (2017). Charge density analysis of metformin chloride, a biguanide anti-hyperglycemic agent. Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials, 73(1), 10-22.
Jelsch, C., Devi, R. N., Noll, B. C., Guillot, B., Samuel, I., & Aubert, E. (2020). Aceclofenac and interactions analysis in the crystal and COX protein active site. Journal of Molecular Structure, 1205, 127600.
Meyer, B., Guillot, B., Ruiz-Lopez, M. F., & Genoni, A. (2016). Libraries of extremely localized molecular orbitals. 1. Model molecules approximation and molecular orbitals transferability. Journal of chemical theory and computation, 12(3), 1052-1067.
Meyer, B., Guillot, B., Ruiz-Lopez, M. F., Jelsch, C., & Genoni, A. (2016). Libraries of extremely localized molecular orbitals. 2. Comparison with the pseudoatoms transferability. Journal of chemical theory and computation, 12(3), 1068-1081.
Howard, E. I., Guillot, B., Blakeley, M. P., Haertlein, M., Moulin, M., Mitschler, A., … & Podjarny, A. (2016). High-resolution neutron and X-ray diffraction room-temperature studies of an H-FABP–oleic acid complex: study of the internal water cluster and ligand binding by a transferred multipolar electron-density distribution. IUCrJ, 3(2), 115-126.
