
At the Interface of Isomorphous Behavior in a 3 × 3 Isomer Grid of Monochlorobenzamides: Analyses of the Interaction Landscapes via Contact Enrichment Studies. Gallagher et al. Cryst. Growth & Design, 19 (2019), 6141-6158.
The physicochemical properties of a 3 × 3 isomer grid of mono-chlorobenzamides (Clxx) are reported with comprehensive studies of their crystal structures and interaction environments (Clx = para-/meta-/ortho-chlorobenzoyl and x = para-/meta-/ortho-aminopyridine substitutions). The nine compound Clxx series was synthesized from the three p-/m-/o-chlorobenzoyl chlorides and three p-/m-/o-aminopyridine isomers using standard synthetic procedures. Clxx exhibits some similarities to the related Fxx and Brxx congeners, e.g., the isomorphous behavior of Clpp (para-chloro-N′-(para-pyridyl)benzamide) with several close relatives, and there are five isomorphous pairs of Clxx and Brxx crystal structures. Notably, Clmp and Clpm both crystallize with Z′ = 4 in space group P1̅ but show important differences. The overall lack of isomers crystallizing with solvate molecules is noteworthy, except for Clmm·(H2O). In all Clxx crystal structures, strong N−H···N hydrogen bonds form; however, Clpo also crystallizes as the unexpected Clpo_O polymorph with N−H···O=C intermolecular hydrogen bonding. The Clxo triad (with ortho-pyridines) exhibits the expected cyclic N−H···N dimer formation with R22(8) hydrogen bonded rings. The HC atom type, forming weak C−H···Cl hydrogen bonds, is the only favored interaction partner of chlorine in Clxx. Conformational analyses (gas phase) together with crystal contact enrichment studies place Clxx in context and at the interface of hydrogen and halogen bonding interactions, though strong hydrogen bonding dominates. In Clxx, the interaction energies with nearest neighbors are shown to contribute to most of the lattice electrostatic energies. The melting temperatures Tm show correlation with both molecular symmetry (Carnelley’s rule) and total electrostatic energy of the weak interactions; in addition, these Tm values can be well predicted from a linear fit combining both descriptors. In Clxx, N−H···N hydrogen bonds dominate, largely in the absence of solvates, and with five Clxx forming isomorphous pairs with Brxx analogues, Clpp being isomorphous with several close benzamide relatives. Analysis of Tm reveals correlations involving both symmetry and electrostatic energies.
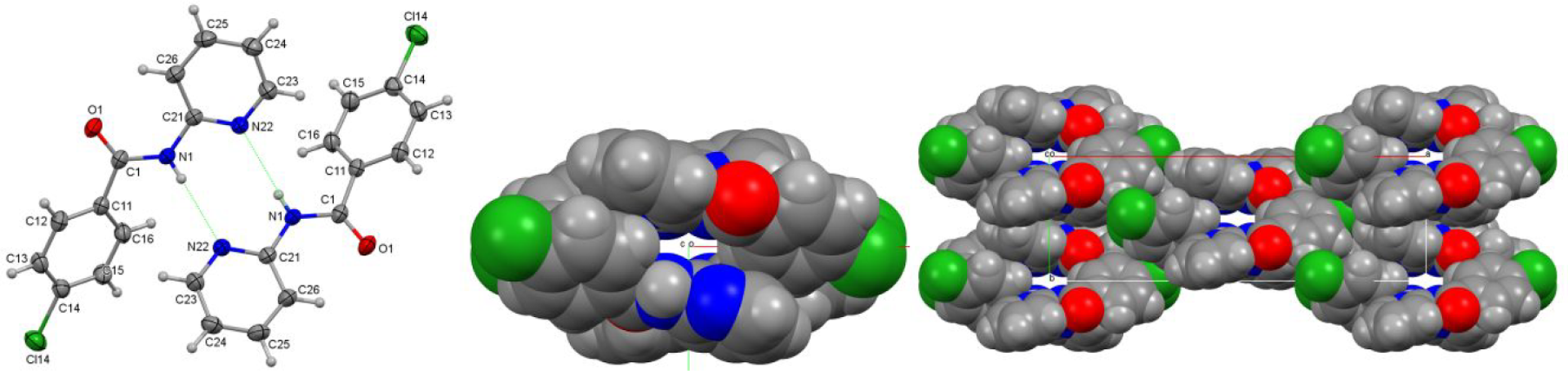
Studies on the relative molecular orientations in the crystalline space and their interaction intensities.
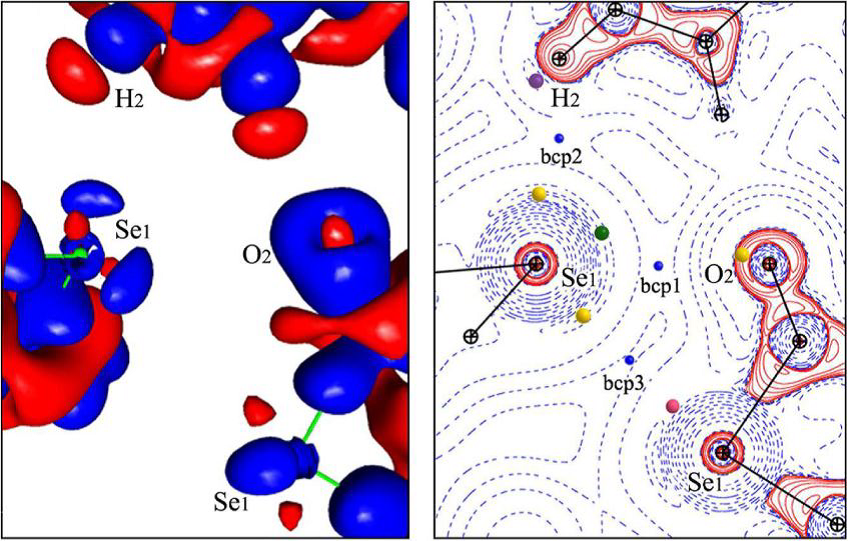
Chalcogen Bonding: Experimental and Theoretical Determinations from Electron Density Analysis. Geometrical Preferences Driven by Electrophilic-Nucleophilic Interactions. Brezgunova et al., Cryst. Growth & Design, 13 (2013), 3283-3289.
The topological analysis of the Laplacian of the electron density ∇2ρ(r) shows that molecular orientation is driven by the interactions between electrophilic and nucleophilic sites found in the external shells of the atoms involved in the intermolecular interactions.
Indeed, this analysis indicates that the relative orientation of three chalcogenated molecules that are assembled in the crystal phase by means of weak interactions is induced by the directions of three electrostatic electrophilic-nucleophilic interactions, which parallel the internuclear directions.
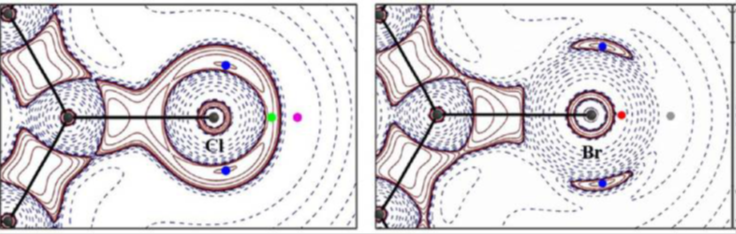
Charge Density Analysis and Topological Properties of Hal3‑Synthons and Their Comparison with Competing Hydrogen Bonds. Brezgunova et al., Cryst. Growth & Design, 12 (2012), 5337-5386.
Trimers based on intermolecular halogen-bonding interactions (Hal3-synthons) have been studied and compared with hydrogen bonds (HBs).
The main differences between Cl3 and Br3-synthons are established coming from the particular structure of the valence shell charge concentration region in Cl and Br atoms. Electrophilic−nucleophilic interactions take place within the intermolecular regions of Hal3-synthons by putting face-to-face charge depletion (CD) and charge concentration (CC) regions belonging to the valence shell of the halogen atoms. The electrostatic interaction CD…CC follows the electrophilic and nucleophilic power of these regions and is monitored by the negative Laplacian values normalized to charge density unit (L/ρ) at the corresponding topological critical points (CPs) of the L(r) = -∇2ρ(r) function. According to the topological and energetic properties at CPs of ρ(r) and L(r), it is observed that Hal3-synthons can successfully compete with intermolecular HBs in the analyzed structures.
Crystalline environment effects on the intensity and nature of intermolecular interactions
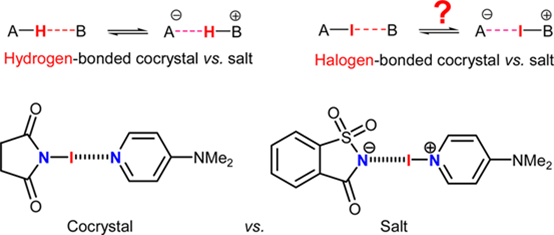
Cocrystal or Salt: Solid State-Controlled Iodine Shift in Crystalline Halogen-Bonded Systems. Makhotkina et al., Cryst. Growth & Design, 15 (2015), 3464-3473.
In the crystal phase any intermolecular interaction is modulated through the crystal electric field generated by the molecular environment.
This effect can be essential for the modification of the original nature of the interaction, allowing a molecular system to migrate from a neutral towards an anionic state. Whereas, in this context, the transfer of a proton is involved in acid-base reactions taking place in molecular solids, this study shows for the first time that the same effect can be achieved with the transfer of a halogen atom.
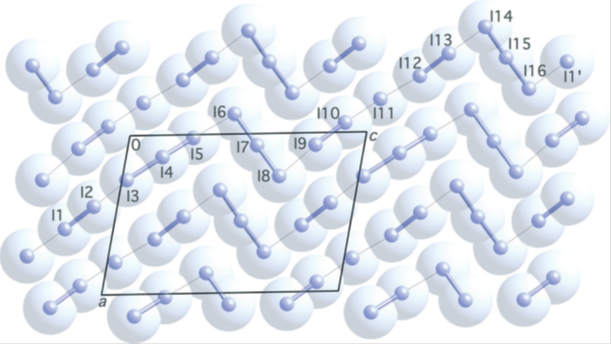
Stabilization of polyiodide chains via anion⋯anion interactions: experiment and theory. Lamberts et al., CrystEngComm, 18 (2016), 3832–3841.
In the crystal structure of tyrosinium polyiodide hydrate, cations and anions aggregate in layers. The anionic part of the structure consists of parallel infinite polyiodide strands; the distance pattern along these chains suggests the presence of smaller subunits I3–, I2 and I–.
Thorough topological analysis carried out with the L = -∇2ρ fonction indicates the successive arrangement of generalized charge concentration and charge depletion sites (for either L > 0 or L < 0 regions) along bond paths, and permits to distinguish iodides from iodine atoms even when they are involved in intermediate situations where interatomic distances and net charges are not conclusive.
Crystal Engineering applied to Material Science : The control of electronic properties of charge transfer materials by the intensity control of halogen bonding between cations and anions
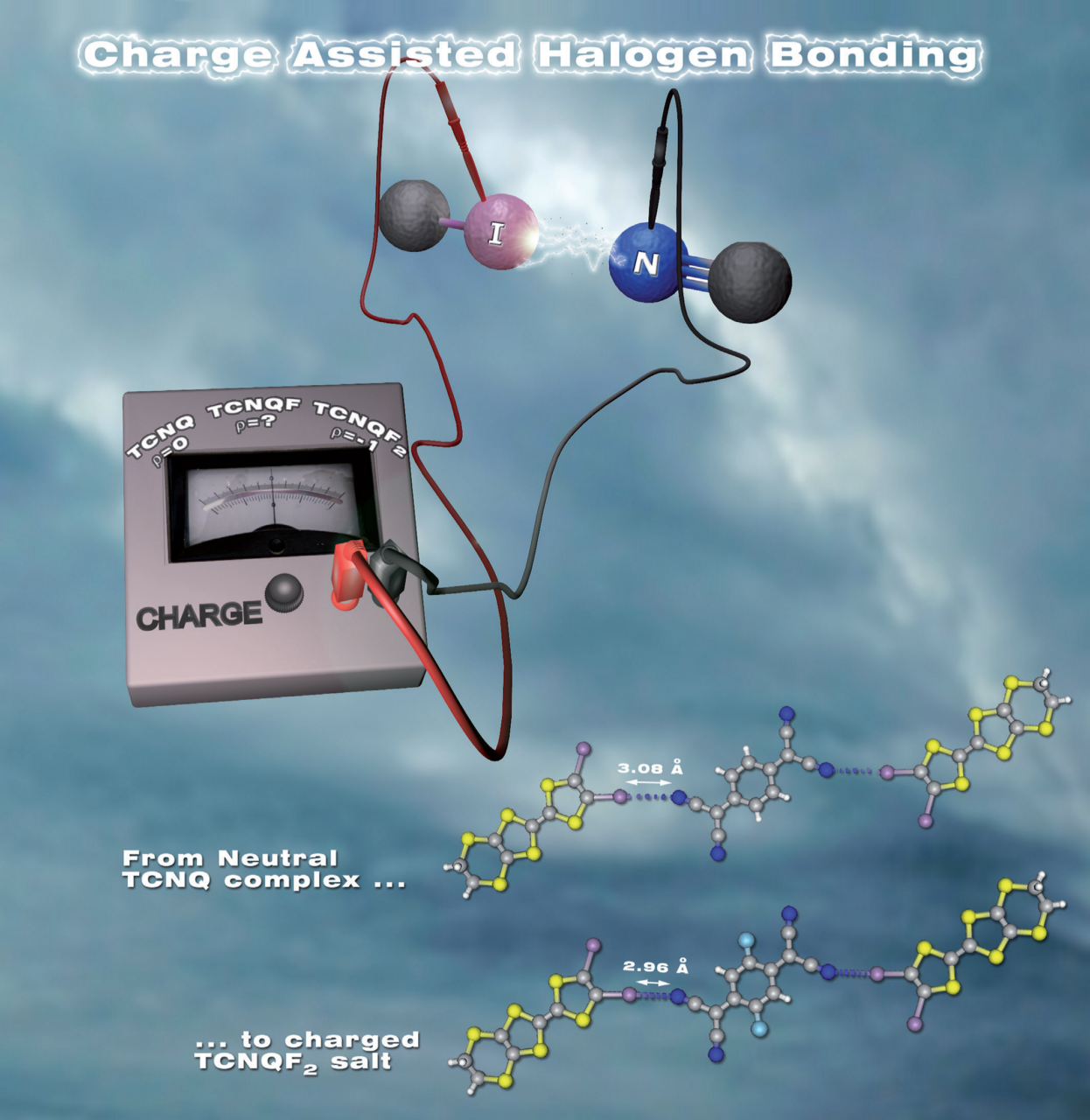
Charge-Assisted Halogen Bonding: Donor-Acceptor Complexes with Variable Ionicity.
Lieffrig et al., Chem. Eur. J., 19 (2013), 14804–14813.
From the structural and electronic studies on a series of isostructural charge-transfer complexes, it has been demonstrated that the degree of charge transfer and the modulation of the intensity of the halogen bond ensuring the assembly are correlated. In the context of Crystal Engineering applied to Material Science, this correlation demonstrates that the control of the intensity of the interaction allows that of the electronic properties of materials constituted by these complexes.
Influence of molecular environment on the properties of crystalline solids of APIs (Active Pharmaceutical Ingredients)
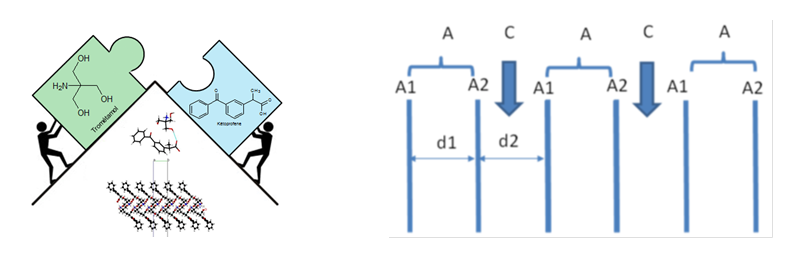
Influence of the crystal environment on the molecular properties of ketoprofene. Ben Nasr et al., in preparation.
Ketoprofen is an anti-inflammatory drug that has a very low water solubility, limiting its bioavailability.
The design and the solubility measurements of several ketoprofen salts, and particularly of racemic ketoprofen-trometamol and two polymorphs of S-ketoprofen-trometamol, confirm that they have greatly improved the solubility compared to the pure active ingredients. The structures of these salts have shown that they are formed by layers of anionic ketoprofenates between which cationic layers are inserted. The study of the thickness of the anionic layers, and the surface and volume occupied by a ketoprofenate anion in a layer, indicates significant variations between the analyzed salts.
The paradox of anionic and cationic aggregates : An electrostatic problem explained in terms of electrophile…nucleophile interactions
Electrostatics at the Origin of the Stability of Phosphate-Phosphate Complexes Locked by Hydrogen Bonds. Mata et al., ChemPhysChem, 13 (2012), 1421–1424.
The phosphate–phosphate interaction presents an intrinsic stability thanks to the formation of hydrogen bonds involving a significant sharing of charge. The electrostatics associated to the formed hydrogen bonds are at the origin of the stability of the complex.
The large energetic barrier observed in the (H2PO4–)2 potential energy surface suggests an important role of interanionic hydrogen bonds in processes involving inter-phosphate aggregation. Thus, once created, the complexes can be stable, prone to survive to changes induced by the environment, either in solution or in solid phase.
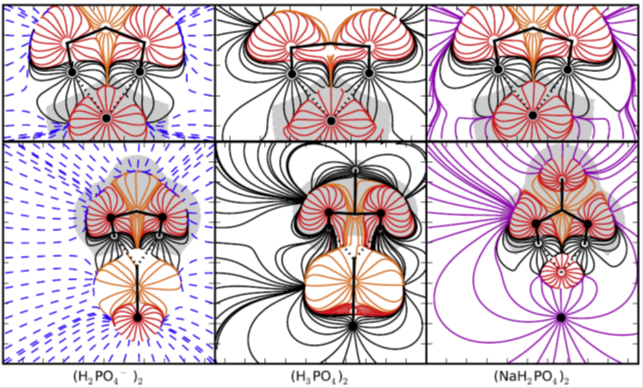
Tracing environment effects that influence the stability of anion–anion complexes: The case of phosphate–phosphate interactions. Mata et al., Chemical Physics Letters, 555 (2013), 106–109.
The effect of the environment on the stability of the (H3PO4)2, (H2PO4–)2, and (HPO42-)2 hydrogen bonded dimers has been explored. The topological analyses of the electron density and the electrostatic potential indicate that he environment has little effect on the hydrogen-bonding interaction, while it induces a significant one on the Coulombic repulsion of the anions.
The interaction energy is represented in terms of hydrogen-bond and non-hydrogen-bond contributions, being only the latter affected by the charge or the environment. While the non-hydrogen bond contribution dominates the interaction energy in the gas phase, it becomes balanced in a polarizable environment.
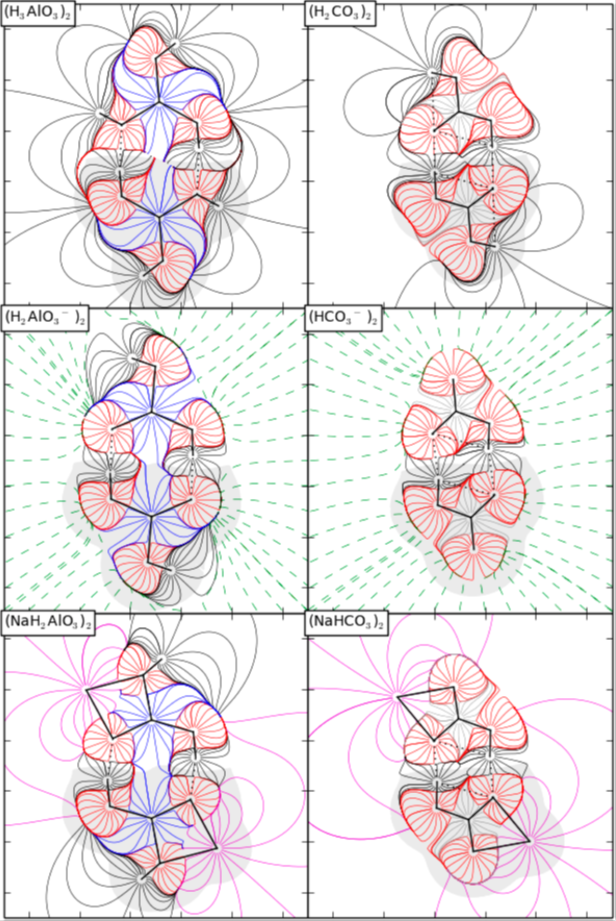
The Paradox of Hydrogen-Bonded Anion−Anion Aggregates in Oxoanions: A Fundamental Electrostatic Problem Explained in Terms of Electrophilic···Nucleophilic Interactions. Mata et al., J. Phys. Chem. A, 119 (2015), 183-194.
The study of anionic complexes formed by two partly deprotonated oxoacids joined by hydrogen bonds has shown that, in spite of the ionic repulsion, local energy minima are found both in gas phase and in aqueous solution. The topologies of the electrostatic potential and the electron density, as well as the comparison with neutral complexes formed by oxoacids, reveal that the ionization has no significant effect on the properties of the hydrogen bonds. The stability of the complexes in the gas phase is explained by attractive forces localized in a volume situated in the hydrogen bond, which is defined as the electrostatic attraction region (EAR) and is determined by the topological analyses of the electron density and the electrostatic potential, as well as the electric field lines.
In solution, the strong anionic repulsion is mostly screened by the effect of the surrounding polar solvent, which only leads to a weak destabilizing interaction in the hydrogen bond region and finally favors the overall stability of the complexes.
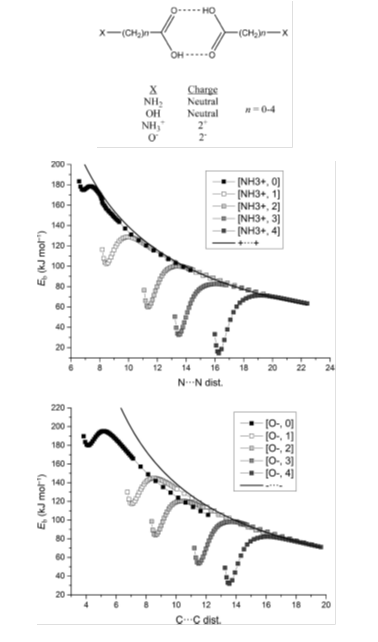
Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Alkorta et al., Chemistry a European Journal, 22 (2016), 9226-9234.
The study of carboxylic acid dimers formed by positively or negatively charged molecules (+2e or -2e), and the corresponding neutral complexes, indicates that no significant differences are found between them. The interaction energy along the dissociation path of the charged dimers shows both a local minimum and a local maximum, defining a stability region in between. When this energetic profile is recalculated by removing the repulsion between the charged groups, it resembles to those of the neutral molecules. The interacting quantum atom (IQA) method shows repulsive electrostatic interactions when the whole molecules are considered in the ionic complexes, but attractive ones between the carboxylic groups in both neutral and ionic complexes.
