

Comparative Analysis of Photoswitching Properties of Analogous Nickel(II) and Palladium(II) Nitrite Complexes in the Solid State
Kacper Paszczyk, Krystyna A. Deresz, Radosław Kamiński, Patryk Borowski, Dominik Schaniel, Adam Krówczyński, Katarzyna N. Jarzembska
Check out the latest photocrystallographic results on molecular photoswitches with our collaborators from Warsaw University, who produced an artistic view for the cover page of Crystal Growth and Design.
Doi : 10.1021/acs.cgd.4c00956 ; HAL: https://hal.science/hal-04964842

La septième édition de l’école de cristallographie mathématique en Amérique Latine rencontre un franc succès au Pérou
Le laboratoire CRM2 continue son investissement dans la formation des jeunes cristallographes dans différentes régions du globe. Cet été, la septième édition de l’école de cristallographie mathématique en Amérique Latine a été organisée à Lima, Pérou. Le Pr Massimo NESPOLO, membre du laboratoire CRM2, était encore une fois parmi les
intervenants et a données plusieurs cours et TD sur la symétrie cristallographiques et les groupes d’espace.
L’agence gouvernementale ProCiencia a publié un compte rendu de l’école et réalisé des interviews avec les intervenants.
Interviews : https://www.facebook.com/share/v/SfciETr7jg7HvsTo/?mibextid=oFDknk
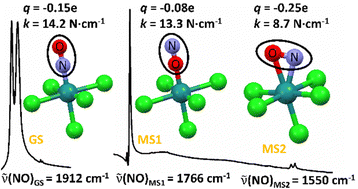
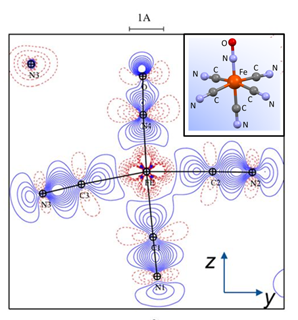
PCCP Hot article: Local force constants and charges of the nitrosyl ligand in photoinduced NO linkage isomers in a prototypical ruthenium nitrosyl complex
Artem A. Mikhailov, Axel Gansmüller, Krzysztof A. Konieczny, Sébastien Pillet, Gennadiy Kostin, Peter Klüfers, Theo Woike and Dominik Schaniel, Phys. Chem. Chem. Phys. 2024,26, 15255-15267
Hot article, https://doi.org/10.1039/D4CP01374C
Photoinduced linkage isomers (PLI) of the NO ligand in transition-metal nitrosyl compounds can be identified by vibrational spectroscopy due to the large shifts of the ν(NO) stretching vibration. Our detailed experimental and theoretical study of the prototypical compound K2[RuCl5NO] shows that the experimentally observed N–O stretching modes in the three linkage isomer configurations GS, MS1, and MS2 exhibit strong coupling with the Ru–N and Ru–O stretching modes, which can be decoupled using the local mode vibrational theory formalism. From the resulting decoupled pure two-atomic harmonic oscillators the local force constants are determined, which all follow the same quadratic behavior on the wavenumber. A Bader charge analysis shows that the total charge on the NO ligand is not correlated to the observed frequency shift of ν(NO).
Pas de Deux of an NO Couple: Synchronous Photoswitching from a Double-Linear to a Double-Bent Ru(NO)2 Core under Nitrosyl Charge Conservation
Asma Hasil, Daniel Beck, Daniel Schröder, Sébastien Pillet, Emmanuel Wenger, Theo Woike, Peter Klüfers, Dominik Schaniel, Angewandte Chemie International Edition 61 (42) e202210671 (2022) https://doi.org/10.1002/anie.202210671
The {Ru(NO)2}10 dinitrosylruthenium complex Ru(NO)2(PPh3)2 shows photo-induced linkage isomerism (PLI) of a special kind: the two NO ligands switch, on photo-excitation, synchronously from the ground state (GS) with two almost linear RuNO functions to a metastable state (MS) which persists up to 230 K and can be populated to ≈50 %. The MS was experimentally characterised by photo-crystallography, IR spectroscopy and DS-calorimetry as a double-bent variant of the double-linear GS. The experimental results are confirmed by computation which unravels the GS/MS transition as a disrotatory synchronous 50° turn of the two nitrosyl ligands. Although 1 shows the usual redshift of the N−O stretch on bending the MNO unit, there is no increased charge transfer from Ru to NO along the GS-to-MS path. In terms of the effective-oxidation-state (EOS) method, both isomers of 1 and the transition state are Ru−II(NO+)2 species.
The animation illustrates the synchronous movement through the transition state (TS) which connects the double-linear ground state (GS) with the double-bent metastable state (MS) of (1). The TS’s strongly negative vibrational mode (−831 cm−1) traces the decay path if the photo-generated MS is warmed above 230 K. 1 provides the first example of synchronous photo-induced linkage isomerism (PLI) involving two ligands
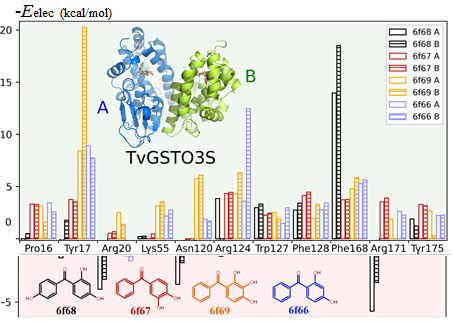
A rush to explore protein–ligand electrostatic interaction energy with Charger.
Vedran Vuković, Theo Leduc, Jelić-Matošević, Claude Didierjean, Frédérique Favier, Benoît Guillot & Christian Jelsch. (2021). Acta Crystallographica D77.
The mutual penetration of electron densities between two interacting molecules complicates the computation of an accurate electrostatic interaction energy. The numerical exact potential and multipole moment (nEP/MM) method is time-consuming since it performs a 3D integration to obtain the electrostatic energy at short interaction distances. A fully analytical computation of the electrostatic interaction energy (aEP/MM) has been reported by Nguyen et al. [2018, Acta Cryst. A74].
Glutathione transferase (GST) in complex with benzophenone ligands was studied due to the availability of both structural and thermodynamic data. The resulting analysis highlights not only the residues that stabilize the ligand but also those that hinder ligand binding from an electrostatic point of view. This offers new perspectives in the search for mutations to improve the interaction between the two partners.
The method performs much faster than nEP/MM (up to two orders of magnitude) and remains highly accurate. A new program library, Charger, contains an implementation of the aEP/MM method. Charger has been incorporated into the MoProViewer software. Benchmark tests on a series of small molecules show the efficiency of Charger in terms of execution time and accuracy. Charger is also powerful in a study of electrostatic symbiosis between a protein and a ligand.
CHARDI2015 : un logiciel pour l’analyse des structures cristallines non-moléculaires

Special Issue: Mathematical Crystallography 2019
Crystal Research and Technology, Volume 55, Issue 5
Guest Editor: Massimo Nespolo
{FeNO}7‐Type Halogenido Nitrosyl Ferrates: Syntheses, Bonding, and Photoinduced Linkage Isomerism
Areenan In‐Iam, Markus Wolf, Claudia Wilfer, Dominik Schaniel, Theo Woike, Peter Klüfers, Chem. Eur. J. 2019, 25, 1304-1325
Our recent article in Chemistry a European Journal concerning the photoinduced isomerism in prototypes of the MNIC and the DNIC classes of nitrosyl complexes has been put into spotlight by Angewandte Chemie Intl. Ed.

Structures on different time scales
Theo Woike, Dominik Schaniel (Eds.) Structures on different time scales March 2018
The book « Structures on different time scales » presents theory and methods to study the structure of condensed matter on different time scales. The authors cover the structure analysis by X-ray diffraction methods from crystalline to amorphous materials, from static-relaxed averaged structures to short-lived electronically excited structures, including detailed descriptions of the time-resolved experimental methods. Complementary, an overview of the theoretical description of condensed matter by static and time-dependent density functional theory is given, starting from the fundamental quantities that can be obtained by these methods through to the recent challenges in the description of time-dependent phenomena such as optical excitations.
With contributions from R. B. Neder, K. Schwarz & P. Blaha, V. Olevano, S. Pillet, B. D. Patterson.
See also book review in Journal of Applied Crystallography by T. Elsaesser,
J. Appl. Cryst. (2019). 52

European Journal of Inorganic Chemistry Volume 2018, Issue 3-4, January 2018
Guest Editors Boris Le Guennic, Guillaume Chastanet, Sébastien Pillet, and Rodolphe Clérac emphasize the pivotal role of Olivier Kahn in the field of molecular magnetism, presenting this issue in honor of his valuable contribution.

Toward a reverse hierarchy of halogen bonding between bromine and iodine
Emmanuel Aubert, Enrique Espinosa, Irène Nicolas, Olivier Jeannin and Marc Fourmigué Faraday Discussions Volume 203, October 2017
We compare here the halogen bond characteristics of bimolecular adducts involving either N-bromo- or N-iodosaccharin as strong halogen bond donors, with 4-picoline as a common XB acceptor. In the NBSac$Pic system, the bromine atom of NBSac is displaced toward the picoline, almost at a median position between the two nitrogen atoms, NSac and N0 Pic, with NSac/Br and Br/N0 Pic distances at 2.073(6) and 2.098(6) °A respectively. This extreme situation contrasts with the analogous iodine derivative, NISac$Pic, where the NSac–I and I/N0 Pic distances amount to 2.223(4) and 2.301(4) °A respectively. Periodic DFT calculations, and molecular calculations of adducts (PBEPBE-D2 aug-cc-pVTZ) either at the experimental frozen geometry or with optimization of the halogen position, indicate a more important degree of covalency (i.e. shared-shell character) in the adduct formed with the bromine atom. A stronger charge transfer to the picoline is also found for the bromine (+0.27 |e|) than for the iodine (+0.18 |e|) system. This inversion of halogen bond strength between I and Br finds its origin in the strong covalent character of the interaction in these adducts, in line with the strength of covalent N–Br and N–I bonds.
The August 2017 special issue pf Acta Crystallographica B on charge density, photocrystallography and time-resolved crystallography: a tribute to Professor Philip Coppens (Guest Editors: Claude Lecomte, Jason Benedict and Yu-Sheng Chen) is dedicated to a lifetime of outstanding scientific achievements by Professor Philip Coppens.
Acta Crystallographica B Special Issue, August 2017
The members of the CRM2 laboratory contributed with various articles in the field of electron density analysis and time-resolved photocrystallography to this issue in remembrance of the impactful scientific legacy of Philip Coppens.
Cover illustration: Images illustrating charge and spin density, and photocrystallography taken from Voufack et al. [(2017). Acta Cryst. B73, 544-549], Mariette et al. [(2017). Acta Cryst. B73, 660-668] and Stachowicz et al. [(2017). Acta Cryst. B73, 643-653].

Keynote Lecture: Crystallographic rationale for the formation of twinned crystals by Massimo Nespolo
Massimo Nespolo IUCr 2017 August 2017
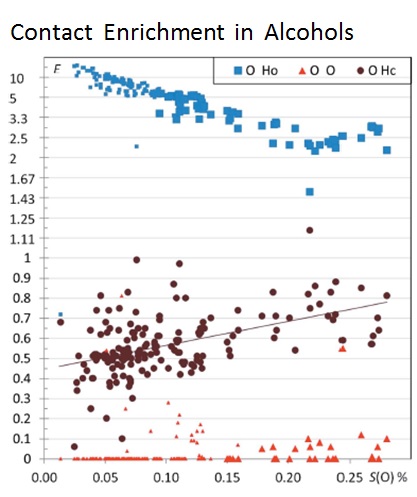
Atom interaction propensities of oxygenated chemical functions in crystal packings
Christian Jelsch & Yvon Bibila Mayaya Bisseyou IUCrJ Volume 4, Part 2, March 2017
The crystal contacts of several families of oxygenated hydrocarbon compounds were analyzed statistically using the Hirshfeld surface methodology. The propensity of contacts to occur between two chemical types is described with the contact enrichment descriptor. The systematic large enrichment ratios of some interactions like the O-H…O hydrogen bonds suggests that these contacts are a driving force in the crystal packing formation. The same statement holds for the weaker C-H…O hydrogen bonds in ethers, esters and ketones, in the absence of polar hydrogen atoms. The over-represented contacts are generally of two types: electrostatic attractions (H-bonds) and hydrophobic interactions. While Cl…O interactions are generally avoided, in a minority of chloro-oxygenated hydrocarbons, significant halogen bonding does occur.
General tendencies can often be derived for many contact types, but outlier compounds are instructive as they display peculiar or rare features. The methodology allows also detecting outliers which can be structures with errors. The behavior of water in monohydrate compounds and of crystals with Z’=2 (dimers) were also investigated. It was found in several cases that cross-interactions between different chemical groups (e.g. water/alcohols; alcohols/phenols) are often favored in the crystal packings. Some unexpected results can also appear. For example, in crystals of alcohol-phenol compounds, the strong O-H…O hydrogen bonds between two phenol groups turn out to be extremely rare, while cross contacts between phenols and alcohols have enriched occurrences.73, 643-653].

Elastic Frustration Triggering Photoinduced Hidden Hysteresis and Multistability in a Two-Dimensional Photoswitchable Hofmann-Like Spin-Crossover Metal–Organic Framework
Eric Milin, Véronique Patinec, Smail Triki, El-Eulmi Bendeif, Sébastien Pillet, Mathieu Marchivie, Guillaume Chastanet, and Kamel Boukheddaden Inorganic Chemistry Volume 55 (22), 11652–11661, July 2016
We report a new two-dimensional Hofmann-like spin-crossover material that experiences strong elastic frustration, leading to an incomplete spin transition. Under light, a hidden stable low-spin state is reached, revealing the existence of a hidden thermal hysteresis and multistability features. These characteristics pave the way for a multidirectional photoswitching and allow potential applications for electronic devices based on ternary digits.

Charge distribution as a tool to investigate structural details. IV. A new route to heteroligand polyhedra
Massimo Nespolo Acta Crystallographica B Volume 72, Issue 1, 51-66, February 2016
The structure of crocoite in the anion-centred description. The Charge Distribution (CHARDI) analysis shows that the structure can be described as centred on cation-centred homoligand polyhedra or anion-centred heteroligand polyhedra.

Special Issue: Mathematical Crystallography / Guest Editors: Massimo Nespolo and Mois Aroyo
Massimo Nespolo Zeitschrift für Kristallographie Volume 230, Issue 13, 2015

Tips and traps on crystal twinning: how to fully describe your twin
Massimo Nespolo Crystal Research and Technology Volume 50, Issue 5, 362–371, May 2015
Derivation of the symmetry of the (011) twin in cassiterite in stereographic projection. H1 and H2 are the symmetry elements of the 4/mmm point group along the a axis for the two individuals, the red line is the twin plane, H* is the intersection group, K is the twin point group obtained as extension of H* by the twin plane.
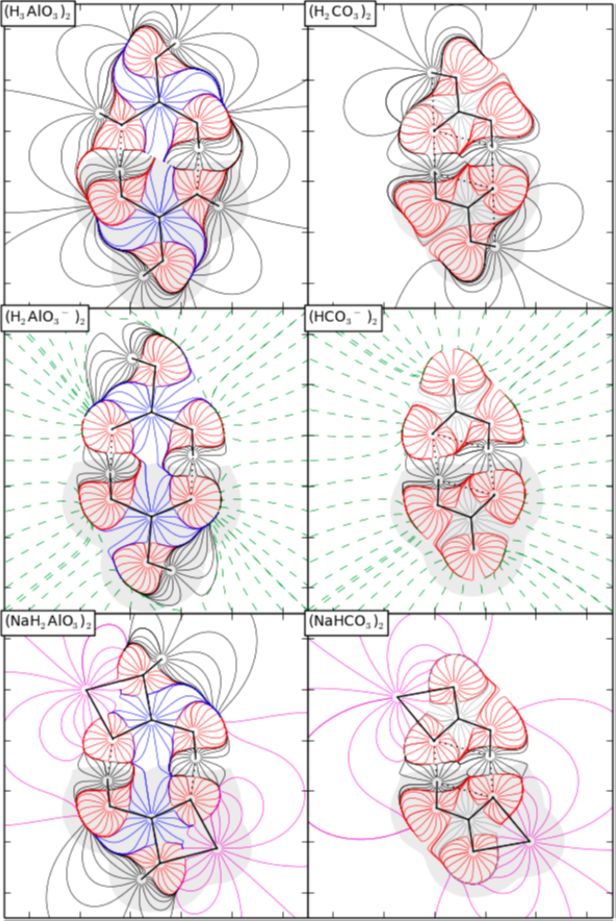
The Paradox of Hydrogen-Bonded Anion−Anion Aggregates in Oxoanions: A Fundamental Electrostatic Problem Explained in Terms of Electrophilic···Nucleophilic Interactions
I. Mata, E. Molins, I. Alkorta, and E. Espinosa, J. Phys. Chem. A (2015),119, 183–194.
Le paradoxe des agrégats anioniques : Un problème électrostatique expliqué à partir des interactions électrophile…nucléophile
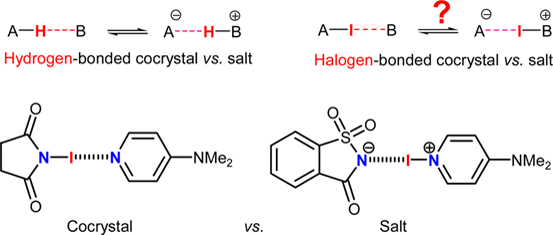
Cocrystal or Salt: Solid State-Controlled Iodine Shift in Crystalline Halogen-Bonded Systems
Makhotkina, O., Lieffrig, J., Jeannin, O., Fourmigué, M., Aubert, E. & Espinosa, E Crystal Growth & Design (2015), 15, 3464-3473
Effet de l’environnement cristallin sur l’intensité et la nature des interactions intermoléculaires
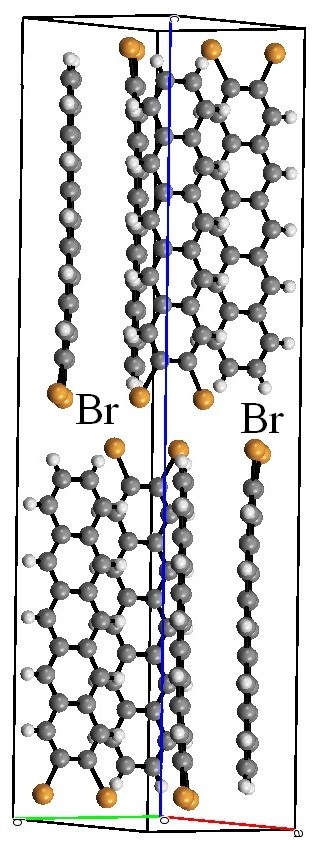
Likelihood of atom-atom contacts in crystal structures of halogenated organic compounds.
Christian Jelsch, Sarra Soudani & Cherif Ben Nasr IUCrJ April 2015 2, 327-340
The likelihood of intermolecular contacts to occur in crystals of halogenated organic compounds was analyzed statistically using tools based on the Hirshfeld surface. The so-called halogen bonding (halogen making short interactions with oxygen or nitrogen, π interaction with carbon) is generally disfavored except when hydrogen is scarce on the molecular surface. Similarly, halogen···halogen contacts are also more rare than expected, except for molecules poor in hydrogen. The hydrogen atom is found to be, in general, the preferred partner of organic halogen atoms in crystal packings. On the other hand, carbon···carbon interactions in parallel π-stacking have a high propensity to occur in halogenated aromatic molecules. The behavior of the four different halogen species is compared in several chemical composition contexts.
The analysis tool can be refined by distinguishing several types for a given chemical species, like hydrogen atoms bound to oxygen and carbon. Such distinction shows, for instance, that C-H···Cl and O-H···O are the preferred synthons in compounds containing both oxygen and chlorine.
![Multiple light-induced NO linkage isomers in the dinitrosyl complex [RuCl(NO)2(PPh3)2]BF4 unravelled by photocrystallographic and IR analysis](https://crm2.univ-lorraine.fr/wp-content/uploads/2022/11/img_pub_h1.png)
Multiple light-induced NO linkage isomers in the dinitrosyl complex [RuCl(NO)2(PPh3)2]BF4 unravelled by photocrystallographic and IR analysis
N. Casaretto, S. Pillet, E.-E. Bendeif, D. Schaniel, A. K. E. Gallien, P. Klüfers and T. Woike, IUCr J., (2015), 2, 35-44
Une photo-isomérisation sélective d’un seul ligand nitrosyl a été caractérisée pour la première fois dans un complexe dinitrosyl, en combinant des expériences de photo-cristallographique et de spectroscopie infrarouge. Plusieurs photo-isomères ont ainsi été détectés, et leur signature spectrale déterminée.
Commentaire scientifique par P. R. Raithby: Photocrystallography reveals new metastable nitrosyl linkage isomers in the solid state. (2015) IUCrJ 2, 5-6
Détecteurs à pixels hybrides : un nouvel outil pour mesurer la structure électronique des solides cristallins
Des physiciens ont réalisé le premier diffractomètre à rayons X de laboratoire mettant en œuvre un détecteur à pixels hybrides. L’amélioration de la sensibilité en intensité qui en résulte permet une reconstitution deux fois plus précise des structures électroniques des solides cristallisés.
XPAD X-ray hybrid pixel detector for charge density quality diffracted intensities on laboratory equipment
E. Wenger, S. Dahaoui, P. Alle, P. Parois, C. Palin, C. Lecomte et D. Schaniel Acta Crystallographica B70, 783-791 (2014)
Commentaire scientifique par D. Stalke: A hybrid pixel detector at an in-house device generating stunning charge density quality data; Acta Cryst. (2014). B70, 781-782
Voir aussi les actualités de la recherche CNRS
Special Issue: Mathematical Crystallography / Guest Editors: Massimo Nespolo and Gregory McColm
Massimo Nespolo Acta Crystallographica Section A Special issue, December 2014
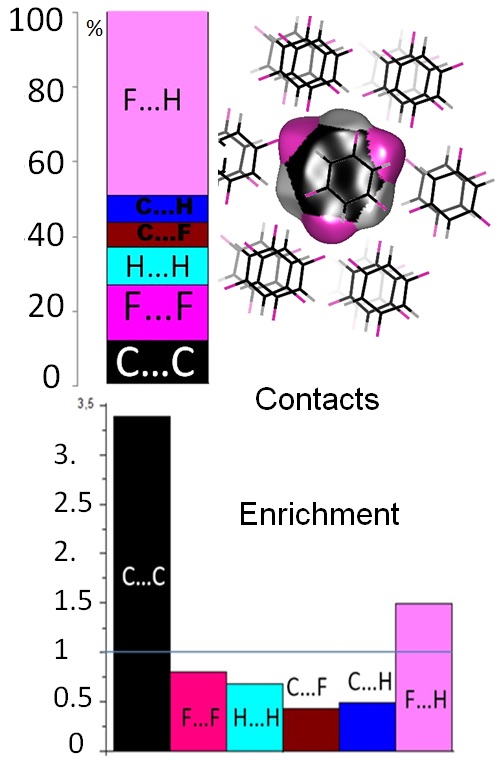
The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis
Christian Jelsch, Krzysztof Ejsmont & Loic Huder IUCrJ February 2014
The crystal contact surface was decomposed into pairs of interacting chemical species to derive an enrichment ratio. This quantity enables the analysis of the propensity of chemical species to form intermolecular interactions with themselves and other species in crystals. The enrichment ratio is obtained by comparing the actual contacts in the crystal with those computed as if all types of contacts had the same probability to form.
As expected, polar contacts of type H…N, H…O, H…S and H…F, which are generally hydrogen bonds, are enriched. O…O and N…N contacts are impoverished while H…H interactions display enrichment ratios which are generally close to unity or slightly lower.
In aromatic compounds, C…C contacts can be very over-represented due to extensive pi…pi stacking in heterocyclic compounds, unlike in pure CH hydrocarbons. Pi…pi stacking is more favourable from an electrostatic point of view in heterocycles than in CH compounds. C…H contacts are favoured in pure CH aromatics, but these interactions occur less in compounds containing O, N or S as some H atoms are then involved in hydrogen bonds.

Room temperature bistability with wide thermal hysteresis in a spin crossover silica nanocomposite
P. Durand, S. Pillet, E.-E. Bendeif, C. Carteret, M. Bouazaoui, H. El Hamzaoui, B. Capoen, L. Salmon, S. Hébert, J. Ghanbaja, L. Aranda and D. Schaniel, J. Mater. Chem. C, (2013), 1, 1901
Une bistabilité à température ambiante a été obtenue pour un nanocomposite élaboré par croissance confinée de nanoparticules à transition de spin [Fe(Htrz)2(trz)](BF4) au sein d’une matrice mésoporeuse de silice de porosité parfaitement contrôlée. Les nanoparticules monodisperses de diamètre 3.5nm présentent l’hystérèse la plus large (65K) jamais atteinte pour des particules de cette taille.

Charge-Assisted Halogen Bonding: Donor-Acceptor Complexes with Variable Ionicity
J. Lieffrig, O. Jeannin, A. Frackowiak, I. Olejniczak, R. Swietlik, S. Dahaoui, E. Aubert, E. Espinosa, P. Auban-Senzier and M. Fourmigué Chemistry: a European Journal (2013), 19, 14804-14813.
Ingénierie Cristalline appliquée à la Science de Matériaux : Le contrôle des propriétés électroniques de composés à transfert de charge par le contrôle de l’intensité de la liaison halogène entre cations et anions.
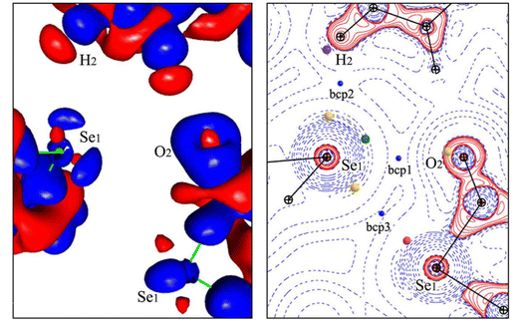
Chalcogen bonding: Experimental and theoretical determinations from electron density analysis. Geometrical preferences driven by electrophilic-nucleophilic interactions
Brezgunova M. E., Lieffrig, J., Aubert, E., Dahaoui, S., Fertey, P., Lebègue, S., Angyan, J. G., Fourmigué, M. & Espinosa, E. Crystal Growth & Design (2013), 13, 3283-3289
Etudes sur l’orientation relative de molécules dans l’espace cristallin et leurs intensités d’interaction
