
The refinement program MoPro (Molecular Properties) is dedicated to the charge-density refinement at (sub)atomic resolution of structures ranging from small molecules to biological macromolecules. MoPro uses the multipolar pseudo-atom model for the electron-density refinement. Alternative methods are also proposed, such as modelling bonding and lone-pair electron density by virtual spherical atoms.
For proteins and organic molecules at atomic resolution, a charge-density database developed in the laboratory enables the transfer of multipolar parameters. The program allows complex refinement strategies to be written and has numerous restraints, constraints and analysis tools for use in the structure and electron-density analysis.
Original kappa, multipolar parameter and stereochemical restraints/constraints are also implemented. Furthermore, constraints on the electron density, such as local symmetry and atom equivalence, are easily defined. Applications range from small molecules to large unit cell crystal structures (enzyme aldose reductase), are given in order to guide the MoPro user and to show the large field of applicability of this code. An automatic charge density refinement procedure is procedure for small molecules at ultra-high resolution (d~0.5Å).
The MoPro program suite is a crystal structure and charge density refinement package which includes two main modules:
– the MoPro least-squares refinement program, which has its own graphical user interface
– VMoPro, a tool dedicated to the computation of properties derived from the electron density.
The software can be download on the BioMod team website, after registration.
MoProViewer is a molecule viewer designed as an interface to VMoPro and is thus especially dedicated to the field of charge density analysis. MoProViewer is written in C++ and is based on the Qt SDK and on the Armadillo and OpenGL libraries. It will be released once published as free of charge and open-source software under the GNU-GPL license.
MoPro and MoProviewer offer compatibility between the MoPro molecular file format (importation and exportation) with other molecular formats (CIF, shelxl, XD, XYZ …).
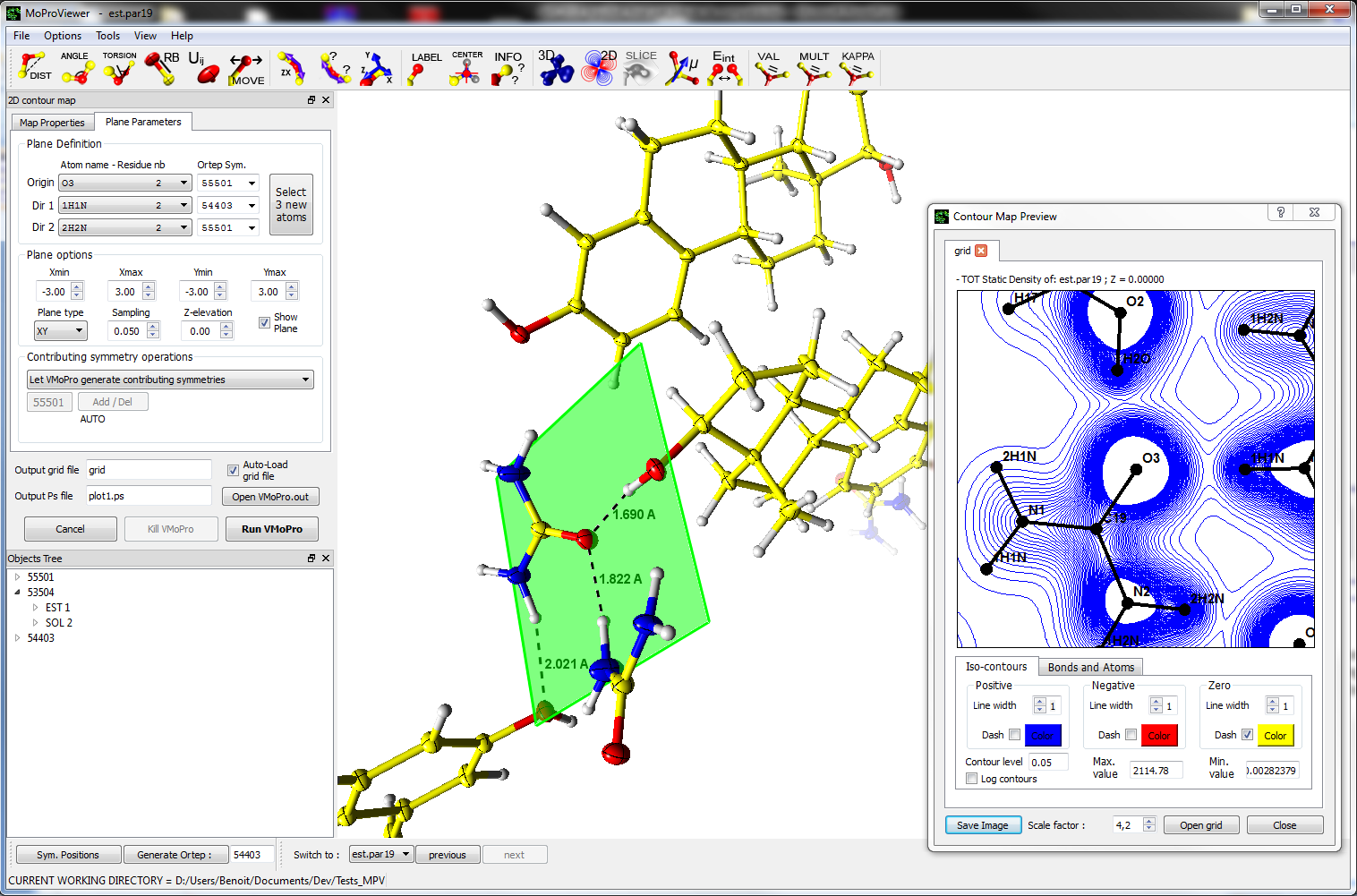
MoProViewer offers a wide range of features, among which:
- Several molecule representation modes (lines, balls &sticks).
- All standard molecule viewer capabilities (configurable atoms labeling and atoms coloring scheme, images exportation).
- All standard crystal structure analysis tools (stereochemistry measurements, symmetry handling,
thermal ellipsoids drawing). - Representation and modification of multipolar model atomic axes systems and chemical equivalency
constraints. - Setup and control of most of the VMoPro possible computations (electron densities, electrostatic potentials, topology …).
- Representation of the molecular properties computed by VMoPro, or readable in XPLOR or Gaussian CUBE format, as 3D isosurfaces, 2D isocontours plots, 3D gradient lines or 2D slices of scalar fields.
- Possibility to color any isosurface on the basis of values of any other loaded scalar field using texture mapping method.
- Drawing of critical points and bond paths obtained from an electron density topology analysis.
- A powerful atom selection tool allowing easy computation focusing on any fragments or regions of a molecule, which is especially convenient in case of large structures (proteins).
- Handling of transferable electron density parameters database.
- Computation and representation of electron density topological basins in the AIM theory
Selection of references :
Vuković, V., Leduc, T., Jelić-Matošević, Z., Didierjean, C., Favier, F., Guillot, B., & Jelsch, C. (2021). Acta Crystallographica Section D: Structural Biology, 77(10). A rush to explore protein–ligand electrostatic interaction energy with Charger.
Leduc, T., Aubert, E., Espinosa, E., Jelsch, C., Iordache, C., & Guillot, B. (2019). The Journal of Physical Chemistry A, 123(32), 7156-7170. Polarization of Electron Density Databases of Transferable Multipolar Atoms.
Fournier, B., Guillot, B., Lecomte, C., Escudero-Adán, E. C., & Jelsch, C. (2018). Acta Crystallographica Section A: Foundations and Advances, 74(3), 170-183. A method to estimate statistical errors of properties derived from charge-density modelling.
Nassour, A., Domagala, S., Guillot, B., Leduc, T., Lecomte, C., & Jelsch, C. (2017). Acta Crystallographica Section B: Structural Science, Crystal Engineering and Materials, 73(4), 610-625. A theoretical-electron-density databank using a model of real and virtual spherical atoms.
Domagala S., Fournier B, Liebschner D., Guillot B. and Jelsch C* Acta Cryst. (2012). A68, 337-351. An improved experimental databank of transferable multipolar atom models – ELMAM2. Construction details and applications.
Guillot. B. Acta Cryst A. (2012). A68, s204. MoProViewer: a molecule viewer for the MoPro charge-density analysis program.
Domagala S. & Jelsch C. (2008) J. Appl. Cryst. 41. Optimal local axes and symmetry assignment for charge-density refinement.
Jelsch, C., Guillot, B., Lagoutte, A. & Lecomte, C. (2005). J. Appl. Cryst. 38, 38-54. Advances in Proteins and Small Molecules. Charge Density Refinement Methods using software MoPro.
